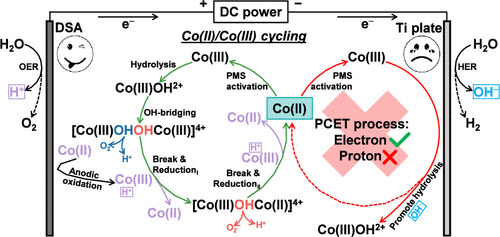
In the realm of transition metal (Mn+) activated peroxymonosulfate (PMS), sluggish reduction kinetics of M(n+1)+ often lead to the rapid deactivation of catalytic centers, posing a significant challenge for commercialization of homogeneous advanced oxidation processes (AOPs). We report a pioneering elucidation of a distinct Co(II)/Co(III) cycling mechanism within electrochemically enhanced PMS-AOPs, utilizing Co(II) as a model catalyst. Remarkably, this cycling process predominantly unfolds in the anodic region, rather than the cathodic, revealing a novel aspect of electrochemical modulation. Co(III), generated by anodic oxidation, emerges as a pivotal species that disrupts the dimerized hydrolysis product ([Co(III)OH]24+). Electron transfer from the hydroxyl oxygen in [Co(III)OH]24+ to Co(III) induces electron redistribution, ultimately facilitating Co(III) reduction and release via both outer- and inner-sphere electron transfer pathways. Gibbs free energy calculations unequivocally confirm the spontaneity of the cyclic process. Our system exhibits superior performance metrics, achieving Co(IV)═O (5.57 × 10–2 mM/M Co) and SO4?– (2.51 × 10–6 mM/M OSO3) yields that surpass most reported catalytic systems, along with an exceptional mass activity of Co(II) (368.87 L/g). This study offers a fresh perspective on Mn+ regeneration for sustained PMS activation in homogeneous transition metal catalysis, with potential implications for advancing the field of environmental remediation.
Article link: https://doi.org/10.1021/acs.est.5c03860

 Address
Address
 E-Mail
E-Mail
 Telephone
Telephone